Abstract:
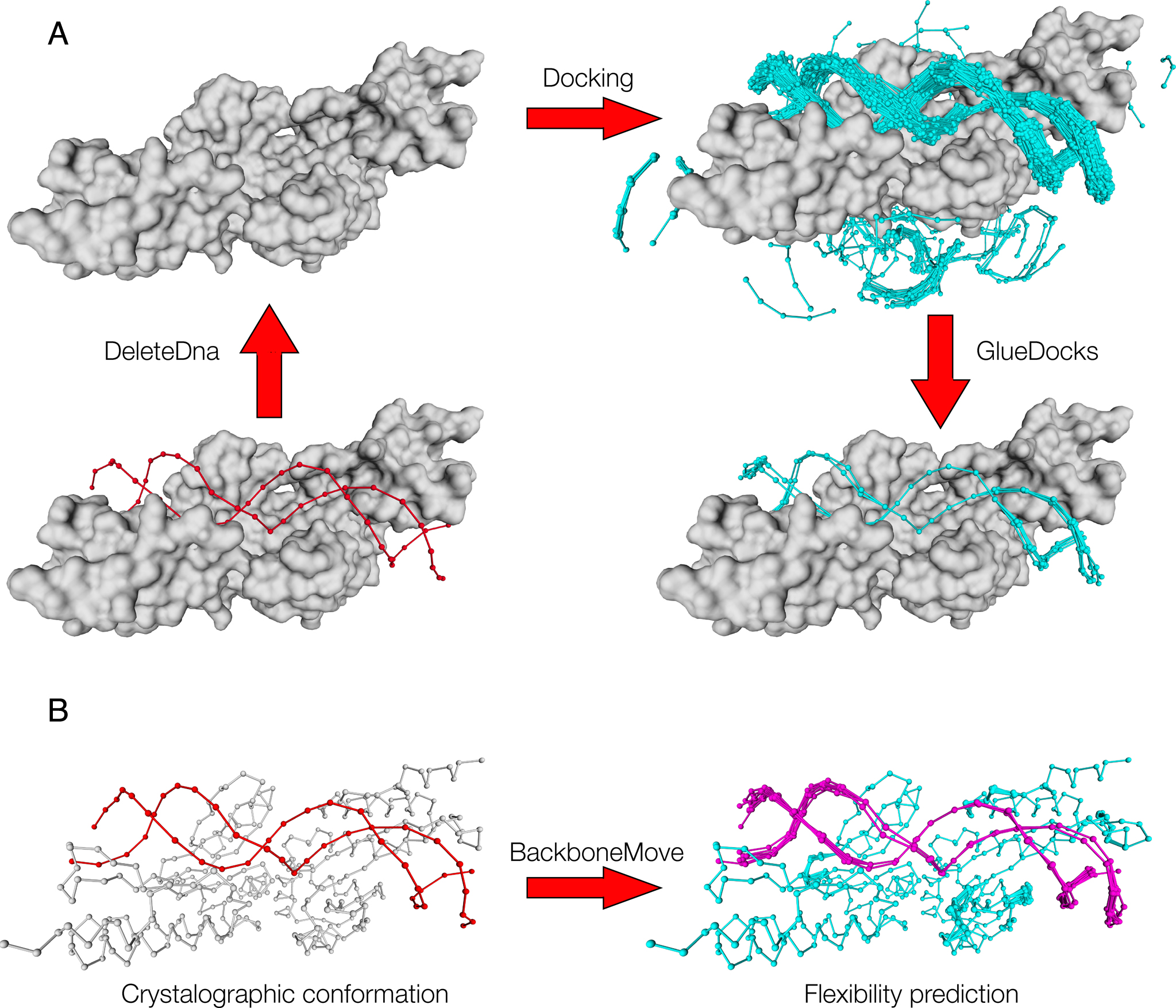
The speed at which new genomes are being sequenced highlights the need for genome-wide methods capable of predicting protein–DNA interactions. Here, we present PADA1, a generic algorithm that accurately models structural complexes and predicts the DNA-binding regions of resolved protein structures. PADA1 relies on a library of protein and double-stranded DNA fragment pairs obtained from a training set of 2103 DNA–protein complexes. It includes a fast statistical force field computed from atom-atom distances, to evaluate and filter the 3D docking models. Using published benchmark validation sets and 212 DNA–protein structures published after 2016 we predicted the DNA-binding regions with an RMSD of <1.8 Å per residue in >95% of the cases. We show that the quality of the docked templates is compatible with FoldX protein design tool suite to identify the crystallized DNA molecule sequence as the most energetically favorable in 80% of the cases. We highlighted the biological potential of PADA1 by reconstituting DNA and protein conformational changes upon protein mutagenesis of a meganuclease and its variants, and by predicting DNA-binding regions and nucleotide sequences in proteins crystallized without DNA. These results opens up new perspectives for the engineering of DNA–protein interfaces.
Read the full text.