Abstract:
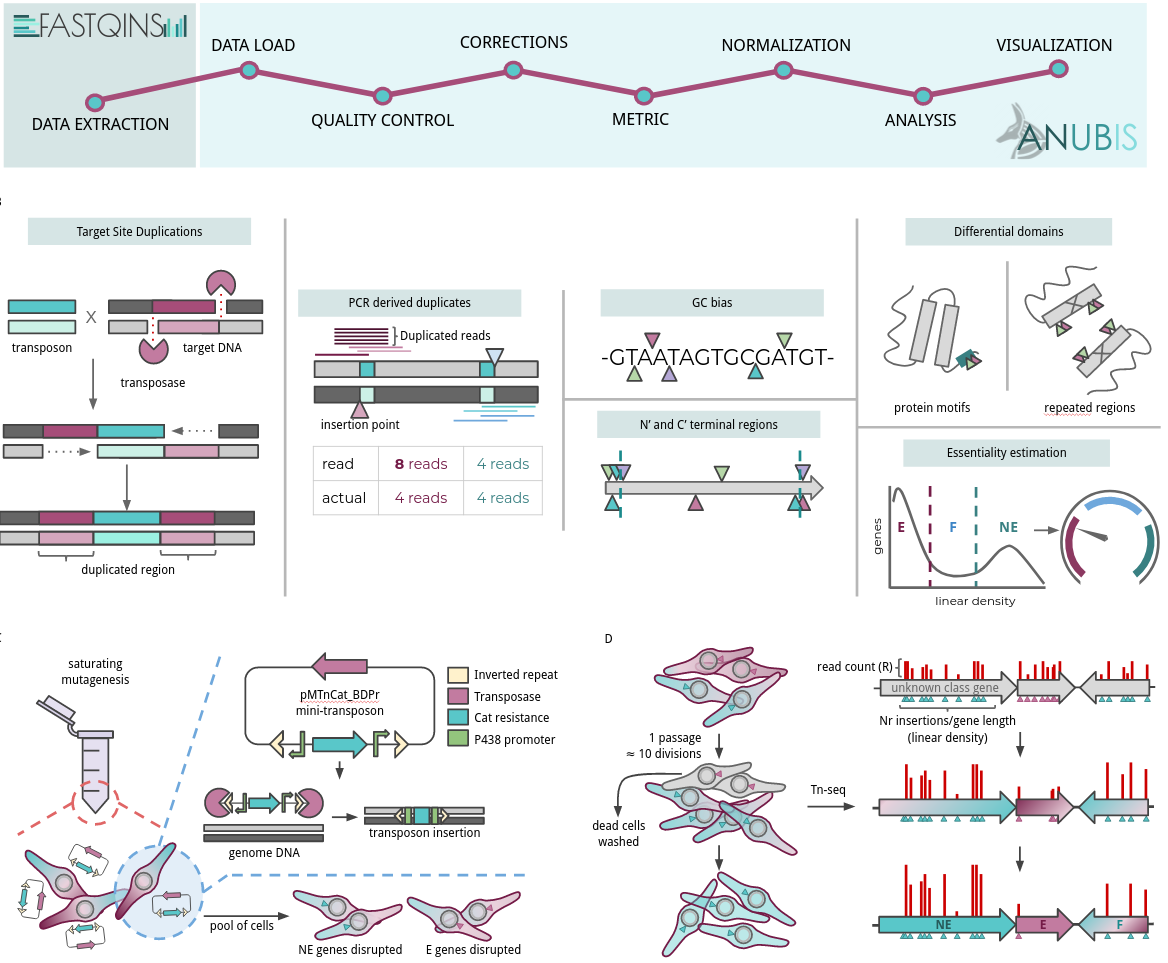
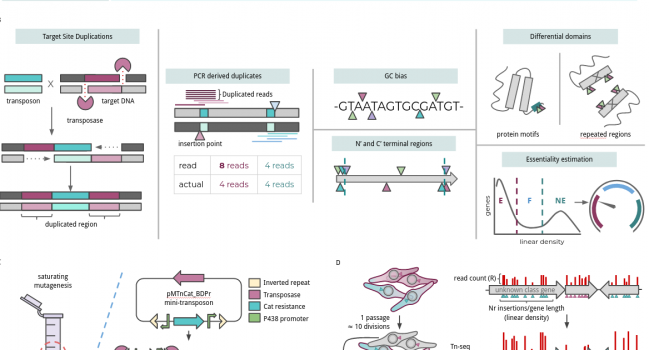
Transposon sequencing is commonly applied for identifying the minimal set of genes required for cellular life; a major challenge in fields such as evolutionary or synthetic biology. However, the scientific community has no standards at the level of processing, treatment, curation and analysis of this kind data. In addition, we lack knowledge about artifactual signals and the requirements a dataset has to satisfy to allow accurate prediction. Here, we have developed FASTQINS, a pipeline for the detection of transposon insertions, and ANUBIS, a library of functions to evaluate and correct deviating factors known and uncharacterized until now. ANUBIS implements previously defined essentiality estimate models in addition to new approaches with advantages like not requiring a training set of genes to predict general essentiality. To highlight the applicability of these tools, and provide a set of recommendations on how to analyze transposon sequencing data, we performed a comprehensive study on artifacts corrections and essentiality estimation at a 1.5-bp resolution, in the genome-reduced bacterium Mycoplasma pneumoniae. We envision FASTQINS and ANUBIS to aid in the analysis of Tn-seq procedures and lead to the development of accurate genome essentiality estimates to guide applications such as designing live vaccines or growth optimization.
Read the full text.